計画研究
中山班は特別推進研究採択のため計画班から離脱し、黒田班の研究協力者になりました。
| A01: 代謝アダプテーション |
2型糖尿病の代謝アダプテーション | 研究代表者 | 黒田 真也(東京大学) |
|---|---|---|---|
| 研究分担者 | 幡野 敦(新潟大学) 土屋 貴穂(筑波大学) |
||
| 研究協力者 | 柚木 克之(理化学研究所) 中山 敬一(九州大学) 松本 雅紀(新潟大学) |
||
| 炎症疾患の代謝アダプテーション | 研究代表者 | 岡田 眞里子(大阪大学) | |
| 研究協力者 | 久保 允人(東京理科大学) | ||
| 薬剤耐性の代謝アダプテーション | 研究代表者 | 松田 史生(大阪大学) | |
| 研究協力者 | 清水 浩(大阪大学) 平井 優美(理化学研究所) 戸谷 吉博(大阪大学) 堀之内 貴明(産業技術総合研究所) |
||
| A02: トランスオミクス解析技術開発 |
次世代メタボローム解析技術開発と応用 | 研究代表者 | 馬場 健史(九州大学) |
| 研究分担者 | 山田 健一(九州大学) | ||
| 研究協力者 | 福崎 英一郎(大阪大学) | ||
| 次世代エピゲノム解析技術の開発とその応用 | 研究代表者 | 伊藤 隆司(九州大学) | |
| 研究分担者 | 梅山 大地(理化学研究所) 荒木 啓充(九州大学) |
||
| 研究協力者 | 三浦 史仁(九州大学) | ||
| 次世代トランスクリプトーム解析技術の開発と応用 | 研究代表者 | 鈴木 穣(東京大学) | |
| 次世代ヒト全ゲノム・オミクスの解析方法論の開発と応用 | 研究代表者 | 角田 達彦(東京大学) | |
| 研究分担者 | 重水 大智(国立長寿医療研究センター) | ||
| 研究協力者 | 宮 冬樹(東京医科歯科大学) |
A01: 代謝アダプテーション
2型糖尿病の代謝アダプテーション

| 研究代表者: | 黒田 真也(東京大学・大学院理学系研究科・教授) |
| 研究分担者: | 幡野 敦(新潟大学・医歯学系・システム生化学分野・助教) 土屋 貴穂(筑波大学・医学医療系 生命科学域 バイオインフォマティクス分野・助教) |
| 研究協力者: | 柚木 克之(理化学研究所・統合生命医科学研究センター・上級研究員) 中山 敬一(九州大学・生体防御医学研究所・主幹教授) 松本 雅記(新潟大学・医歯学系・分子遺伝学分野・教授) |
HPhttp://kurodalab.bs.s.u-tokyo.ac.jp/ja/top/
概要
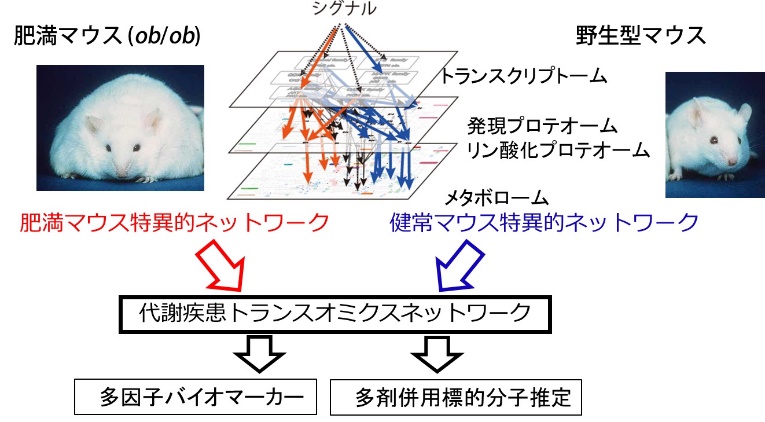
野生型および肥満を伴う2型糖尿病のモデルマウスとして知られる肥満モデルマウス(高脂肪食摂取マウスやob/obマウス)を用いて糖負荷に対する代謝アダプテーションとその破たんを対象にトランスオミクス解析を行う。野生型および肥満モデルマウスに対して、糖負荷前後の肝臓、筋肉、脂肪細胞のメタボローム(馬場)、プロテオーム(中山)、トランスクリプトーム(鈴木)、エピゲノム(伊藤)の時系列を同一個体からのトランスオミクスデータとして取得する。トランスオミクスデータから事前知識を利用して糖負荷による代謝制御システムのトランスオミクスネットワークを同定する。野生型および肥満モデルマウスのトランスオミクスネットワークの差分から、疾患特異的に出現または欠損しているネットワークを同定する。統計的なアプローチを用いて代謝疾患の多因子バイオマーカーを選択する(角田)。酵素反応をベースとした代謝適応システムの数理モデルを作成して、感受性解析から代謝異常を復元できる分子の組み合わせを同定して多剤併用療法の多因子標的の候補を推定する。
参考文献
- Sano, T., Kawata, K., Ohno, S., Yugi, K., Kakuda, H., Kubota, H., Uda, S., Fujii, M., Kunida, K., Hoshino, D., Hatano, A., Ito, Y., Sato, M., Suzuki, Y., & Kuroda, S. Selective control of up-regulated and down-regulated genes by temporal patterns and doses of insulin. Science signaling 9, ra112 (2016).
- Yugi, K., Kubota, H., Hatano, A. & Kuroda, S. Trans-Omics: How To Reconstruct Biochemical Networks Across Multiple 'Omic' Layers. Trends in biotechnology 34, 276-290 (2016).
- Yugi, K., Kubota, H., Toyoshima, Y., Noguchi, R., Kawata, K., Komori, Y., Uda, S., Kunida, K., Tomizawa, Y., Funato, Y., Miki, H., Matsumoto, M., Nakayama, K. I., Kashikura, K., Endo, K., Ikeda, K., Soga, T. & Kuroda, S. Reconstruction of insulin signal flow from phosphoproteome and metabolome data. Cell reports 8, 1171-1183 (2014).

A01: 代謝アダプテーション
炎症疾患の代謝アダプテーション
 研究代表者:岡田 眞里子(大阪大学・蛋白質研究所・教授)
研究代表者:岡田 眞里子(大阪大学・蛋白質研究所・教授)
研究協力者:久保 允人(東京理科大学・生命科学研究所・教授)
HPhttp://www.protein.osaka-u.ac.jp/cell_systems/
概要
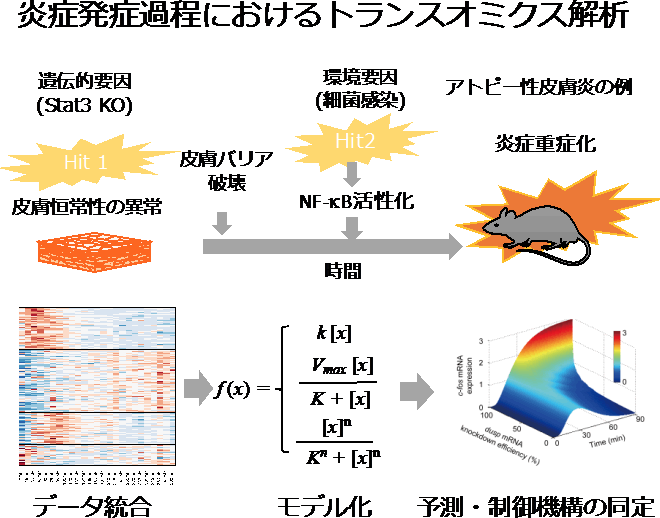
細胞は環境の情報を集約し、遺伝子発現や代謝の活性化を介して、細胞の恒常性の維持と適応を行っている。免疫システムにおいては、シグナル伝達、転写、翻訳、代謝の制御に免疫細胞間の相互作用が中心的役割を果たし、高次のネットワークの秩序を保っている。さらに、最近の研究では、この免疫システム制御において、周辺組織細胞と免疫細胞の相互作用が重要であることも指摘されている。本研究は、アトピー性皮膚炎をはじめとした慢性炎症、免疫応答、免疫応答脱制御に関わる細胞や組織におけるトランスクリプトーム、エピゲノム、プロテオーム、メタボロームのトランスオミクス解析を行い、免疫細胞や周辺細胞の相互作用における代謝の役割を明らかにする。オミクスデータから、代謝制御の実体を担う転写因子を計算的に予測し、転写因子を起点として、さまざまな時間発展あるいは疾病の重篤度に依存したデータ統合を行い、トランスオミクスネットワークの再構成を行う。
数理モデルを用いた再構成ネットワークのシミュレーション解析を通じて、免疫トランスオミクスネットワークにおける代謝アダプテーションの制御機構の理解と疾患操作を目指す。
参考文献
- Inoue, K., Shinohara, H., Behar, M., Yumoto, N., Tanaka, G., Hoffmann, A., Aihara, K. & Okada-Hatakeyama M. Oscillation dynamics underlies functional switching of NF-κB for B cell activation. npj Systems Biology & Application in print.
- Shinohara, H., Behar, M., Inoue, K., Hiroshima, M., Yasuda, T., Nagashima, T., Kimura, S., Sanjo, H., Maeda, S., Yumoto, N., Ki, S., Akira, S., Sako, Y., Hoffmann, A., Kurosaki, T. & Okada-Hatakeyama, M. Positive feedback within a kinase signaling complex functions as a switch mechanism for NF-kappaB activation. Science 344, 760-764 (2014).
- Nakakuki, T., Birtwistle, M.R., Saeki, Y., Yumoto, N., Ide, K., Nagashima, T., Brusch, L., Ogunnaike, B.A., Okada-Hatakeyama, M. & Kholodenko, B.N. Ligand-specific c-Fos expression emerges from the spatiotemporal control of ErbB network dynamics. Cell 141, 884-896 (2010)
A01: 代謝アダプテーション
薬剤耐性の代謝アダプテーション

| 研究代表者: | 松田 史生(大阪大学・大学院情報科学研究科・教授) |
| 研究協力者: | 清水 浩(大阪大学・大学院情報科学研究科・教授) 平井 優美(理化学研究所・環境資源科学研究センター・チームリーダー) 戸谷 吉博(大阪大学・大学院情報科学研究科・助教) 堀之内 貴明(産業技術総合研究所・人工知能研究センター・主任研究員) |
HPhttp://www-symbio.ist.osaka-u.ac.jp/
概要
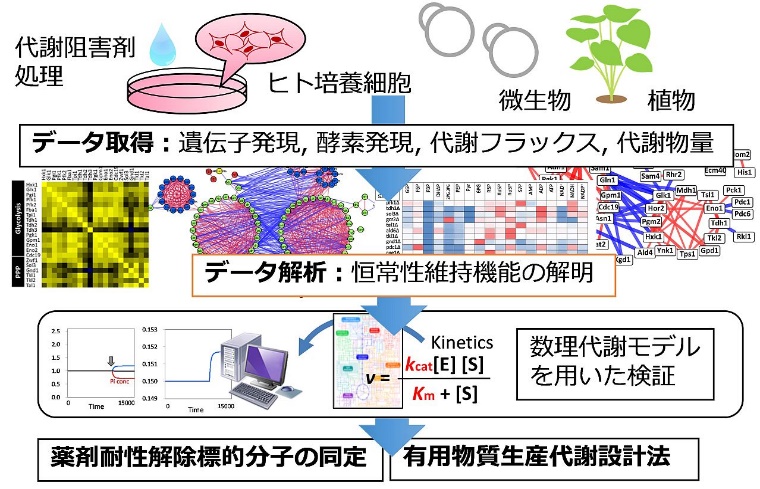
薬剤投与を受けたヒト培養細胞が代謝適応する過程のトランスオミクス解析を行い、薬剤耐性の発現機構を明らかにする。近年代謝酵素を標的とした薬剤の開発が進んでいるが、代謝阻害を受けた細胞は、その影響を打ち消すような代謝アダプテーションを起こして薬剤耐性を示すと考えられている。そこで呼吸阻害剤、解糖系阻害剤等を投与したヒト培養細胞から、メタボローム(馬場)、プロテオーム(中山)、トランスクリプトーム(鈴木)、エピゲノム(伊藤)および数理代謝モデルの構築に必須の代謝フラックス情報(戸谷)を取得する。階層をつないだデータ解析を行い、薬剤耐性に関わるトランスオミクスネットワークを同定し、恒常性維持機構を解明する(松田)。得られたデータから数理代謝モデルを構築し、アロステリック制御、翻訳後制御、酵素発現量制御の検証と、薬剤耐性解除の標的分子の同定を行う。また、代謝遺伝子を欠損した微生物(清水)や代謝に摂動を与えた植物(平井)においても同様の解析を行い、微生物中心代謝、植物アミノ酸代謝の恒常性維持機構を解明するとともに、有用物質生産代謝設計法の開発につなげる。
参考文献
- Matsuda, F., Kinoshita, S., Nishino, S., Tomita, A., & Shimizu, H. Targeted proteome analysis of single-gene deletion strains of Saccharomyces cerevisiae lacking enzymes in the central carbon metabolism. PLoS ONE 12, e0172742 (2017).
- Maeda, K., Okahashi, N., Toya, Y., Matsuda, F. & Shimizu, H. Investigation of useful carbon tracers for 13C-metabolic flux analysis of Escherichia coli by considering five experimentally determined flux distributions. Metabolic engineering communications 3, 187-195 (2016).
- Nakajima, T., Kajihata, S., Yoshikawa, K., Matsuda, F., Furusawa, C., Hirasawa, T. & Shimizu, H. Integrated Metabolic Flux and Omics Analysis of Synechocystis sp. PCC 6803 under Mixotrophic and Photoheterotrophic Conditions. Plant & cell physiology(2014).
A02: トランスオミクス解析技術開発
次世代メタボローム解析技術開発と応用

| 研究代表者: | 馬場 健史(九州大学・生体防御医学研究所・教授) |
| 研究分担者: | 山田 健一(九州大学・大学院薬学研究院・教授 |
| 研究協力者: | 福崎 英一郎(大阪大学・大学院工学研究科・教授) |
HPhttp://bamba-lab.com/?lang=ja
概要
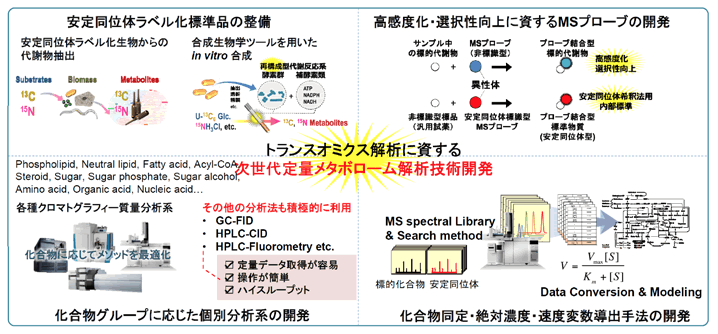
代謝アダプテーションを理解するためには、外乱に対する代謝ネットワークの再構成過程を表現型や多階層オミクスデータと対応させながら経時的に追跡する必要がある。このためには代謝ネットワークを構成する変数である化合物濃度と酵素濃度を精密に測定する必要がある。現状、一般的に用いられるクロマトグラフィー質量分析では、化合物濃度の絶対定量に際し安定同位体ラベル化体が必要になるが、標準品入手等の問題から絶対定量値の取得は困難な状況である。代謝アダプテーション解析に資する次世代メタボロミクス解析基盤を構築するため、馬場班では①定量解析用安定同位体ラベル化標準品のin vivo、in vitro合成(馬場、山田、和泉、相馬)②化合物グループ個別定量分析系の開発(馬場、和泉、相馬)、③安定同位体標識化物を含む複雑なメタボロミクス生データから成分の同定、絶対定量値、速度パラメータを高速に導出可能なデータ解析手法の開発に取り組む(相馬)。これらメタボローム解析基盤と定量プロテーム解析技術(中山)を軸として他オミクス解析グループ[トランスクリプトミクス(鈴木)、エピゲノム(伊藤)とも連携をしながら、代謝アダプテーションに関する応用研究に取り組む(馬場、相馬)。また、開発したメタボローム解析技術については、A01(黒田、中山、岡田、松田)や公募班へ提供し共有する。
参考文献
- Takeda, H., Koike, T., Izumi, Y., Yamada, T., Yoshida, M., Shiomi, M., Fukusaki, E. & Bamba, T. Lipidomic analysis of plasma lipoprotein fractions in myocardial infarction-prone rabbits. Journal of bioscience and bioengineering 120, 476-482 (2015).
- Taguchi, K., Fukusaki, E. & Bamba, T. Simultaneous analysis for water- and fat-soluble vitamins by a novel single chromatography technique unifying supercritical fluid chromatography and liquid chromatography. Journal of chromatography. A 1362, 270-277 (2014).
- Yamada, T., Uchikata, T., Sakamoto, S., Yokoi, Y., Nishiumi, S., Yoshida, M., Fukusaki, E. & Bamba, T. Supercritical fluid chromatography/Orbitrap mass spectrometry based lipidomics platform coupled with automated lipid identification software for accurate lipid profiling. Journal of chromatography. A 1301, 237-242 (2013).
A02: トランスオミクス解析技術開発
次世代エピゲノム解析技術の開発とその応用
 研究代表者:伊藤 隆司(九州大学・大学院医学研究院・教授)
研究代表者:伊藤 隆司(九州大学・大学院医学研究院・教授)
研究分担者:梅山 大地(理化学研究所・統合生命医科学研究センター・研究員)
研究協力者:三浦 史仁(九州大学・大学院医学研究院・講師)
HPhttps://www.biochem1.med.kyushu-u.ac.jp/
概要
多階層計測に基づくトランスオミクスでは、各階層の計測を微量試料から実現することと、単一の計測で多種多様な情報を同時取得することの2点が重要になる。本課題においては、この2つの観点からトランスオミクスに適した2種類の次世代エピゲノム解析技術を開発する。
①メチローム解析:我々は世界最高感度の全ゲノムおよび標的バイサルファイトシーケシング技術 PBAT-seqを開発した。本課題では、単一細胞PBAT-seqの高度化を進めてゲノム被覆率を向上させる一方、合成ロングリードによる二倍体メチローム解読法の開発にも取り組み、メチローム解析の次世代化を図る。②ゲノムフットプリント解析:対象とする核内因子毎に実験を行うChIP-Seqでは、DNA-核内因子相互作用の全貌解明は不可能である。我々は、膜透過性のジメチル硫酸(DMS)を生細胞に直接作用させてin vivoゲノムットプリントを取得する新技術DMS-seqを開発した。DMS-seqは、ヌクレオソームも含めて、あらゆる核内因子の結合をピークとして検出できる。各ピークに予めアノテーションが付与されていると、データ解釈が容易になって、DMS-seqの利便性が飛躍的に高まる。そこで本課題では、このようなinformation-based DMS-seq(iDMS-seq)システムを、まず酵母をモデルに構築し、哺乳類へと拡張する。これらの独自技術を中心に様々なエピゲノム解析技術の開発・改良に取り組む。そうして確立された技術をオンコメタボライトに対するエピゲノム応答の解析に応用するとともに、肥満モデルマウス(黒田)や炎症モデルマウス(岡田)の時系列解析、がん代謝シフトの解析(中山)、免疫薬剤や遺伝子破壊への応答の解析(松田)に提供する。
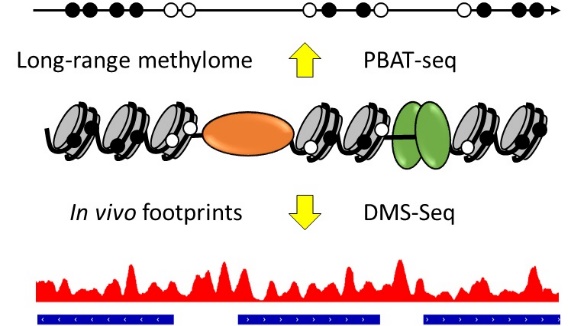
参考文献
- Umeyama, T. & Ito, T. DMS-seq for in vivo genome-wide mapping of protein-DNA interactions and nucleosome centers. Cell reports 21, 289-300 (2017).
- Miura, F. & Ito, T. Highly sensitive targeted methylome sequencing by post-bisulfite adaptor tagging. DNA research 22, 13-18 (2015).
- Miura, F., Enomoto, Y., Dairiki, R. & Ito, T. Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging. Nucleic acids research 40, e136 (2012).
A02: トランスオミクス解析技術開発
次世代トランスクリプトーム解析技術の開発と応用
 研究代表者:鈴木 穣(東京大学・新領域創成科学研究科・教授)
研究代表者:鈴木 穣(東京大学・新領域創成科学研究科・教授)
HPhttp://www.cb.k.u-tokyo.ac.jp/suzukilab/
概要
 本研究課題では、本研究領域のすべての研究班に対して、次世代シークエンスプラットフォームを提供する。同時に本研究班でも独自に、がん細胞の薬剤応答時の代謝アダプテーションにおける多様性を解明するための基盤技術を開発、その実践を行う。これまでに申請者は、代表的な肺腺がん培養細胞株26種について、ゲノム、トランスクリプトーム、エピゲノムのマルチオミクスカタログ化を完了しているが、この細胞株パネルに対して直接の薬剤介入である代謝阻害剤あるいは種々のエピゲノム試薬を投与し、同様にトランスクリプトーム、エピゲノム解析を行う。解析の高効率化が必須になるが、これにはFluidigm社C1システムを反応容器とした用いることで、数百のRNA-seq、ATAC-seqライブラリーを構築することで対応が可能であることを予備的検討から確認している。今回、26細胞株に96薬剤を添加した約2500のライブラリーの構築とシークエンス解析が可能であるかを検証、実践する。また微視的視野から、特に薬剤摂動時の細胞集団内トランスクリプトーム応答の多様性を解析することを目的に、上記技術のシングルセル解析への拡充を試みる。シングルセルの単離には10X Genomics社の提供するChromiumシステムを援用したバーコーディング技術を駆使する。これらの技術をA01(黒田、中山、岡田、松田)や公募班へ提供する。
本研究課題では、本研究領域のすべての研究班に対して、次世代シークエンスプラットフォームを提供する。同時に本研究班でも独自に、がん細胞の薬剤応答時の代謝アダプテーションにおける多様性を解明するための基盤技術を開発、その実践を行う。これまでに申請者は、代表的な肺腺がん培養細胞株26種について、ゲノム、トランスクリプトーム、エピゲノムのマルチオミクスカタログ化を完了しているが、この細胞株パネルに対して直接の薬剤介入である代謝阻害剤あるいは種々のエピゲノム試薬を投与し、同様にトランスクリプトーム、エピゲノム解析を行う。解析の高効率化が必須になるが、これにはFluidigm社C1システムを反応容器とした用いることで、数百のRNA-seq、ATAC-seqライブラリーを構築することで対応が可能であることを予備的検討から確認している。今回、26細胞株に96薬剤を添加した約2500のライブラリーの構築とシークエンス解析が可能であるかを検証、実践する。また微視的視野から、特に薬剤摂動時の細胞集団内トランスクリプトーム応答の多様性を解析することを目的に、上記技術のシングルセル解析への拡充を試みる。シングルセルの単離には10X Genomics社の提供するChromiumシステムを援用したバーコーディング技術を駆使する。これらの技術をA01(黒田、中山、岡田、松田)や公募班へ提供する。
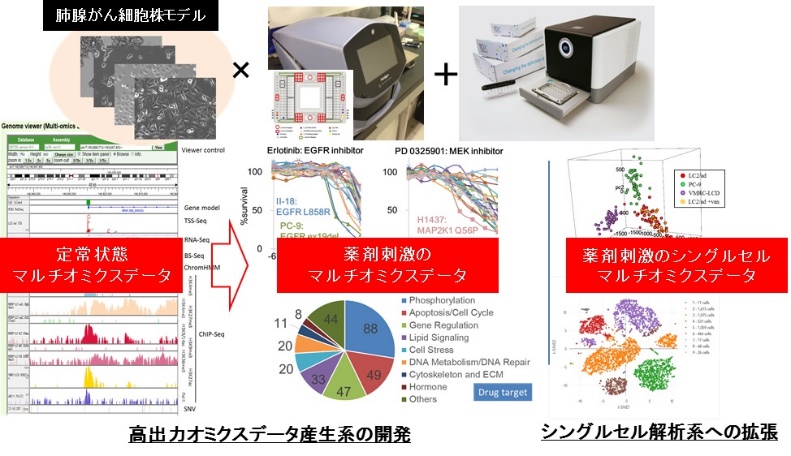
参考文献
- Hashimoto, T., Horikawa, D. D., Saito, Y., Kuwahara, H., Kozuka-Hata, H., Shin, I. T., Minakuchi, Y., Ohishi, K., Motoyama, A., Aizu, T., Enomoto, A., Kondo, K., Tanaka, S., Hara, Y., Koshikawa, S., Sagara, H., Miura, T., Yokobori, S., Miyagawa, K., Suzuki, Y., Kubo, T., Oyama, M., Kohara, Y., Fujiyama, A., Arakawa, K., Katayama, T., Toyoda, A. & Kunieda, T. Extremotolerant tardigrade genome and improved radiotolerance of human cultured cells by tardigrade-unique protein. Nature communications 7, 12808 (2016).
- Suzuki, A., Matsushima, K., Makinoshima, H., Sugano, S., Kohno, T., Tsuchihara, K. & Suzuki, Y. Single-cell analysis of lung adenocarcinoma cell lines reveals diverse expression patterns of individual cells invoked by a molecular target drug treatment. Genome biology 16, 66 (2015).
- Mino, T., Murakawa, Y., Fukao, A., Vandenbon, A., Wessels, H. H., Ori, D., Uehata, T., Tartey, S., Akira, S., Suzuki, Y., Vinuesa, C. G., Ohler, U., Standley, D. M., Landthaler, M., Fujiwara, T. & Takeuchi, O. Regnase-1 and Roquin Regulate a Common Element in Inflammatory mRNAs by Spatiotemporally Distinct Mechanisms. Cell 161, 1058-1073 (2015).
A02: トランスオミクス解析技術開発
次世代ヒト全ゲノム・オミクスの解析方法論の開発と応用
 研究代表者:角田 達彦(東京大学・大学院理学系研究科・教授)
研究代表者:角田 達彦(東京大学・大学院理学系研究科・教授)
研究分担者:重水 大智(国立長寿医療研究センター・臨床ゲノム解析推進部・ユニット長)
研究協力者:宮 冬樹(東京医科歯科大学・難治疾患研究所・助教)
概要
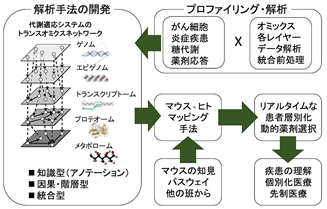 本計画研究では、トランスオミクス解析の手法の提案・開発、ヒトのデータを中心とした解析、そしてマウスの知見をヒトへマッピング・変換、共通する機序を探ることを主軸とする。まず、トランスオミクス解析の方法論として、知識型、因果・階層型、統合型の3種類の方法を検討する。知識型として、染色体構造などのゲノムアノテーションデータにより、ゲノムから代謝にいたる制約を見出すことでフィルタリングを行う。因果・階層型として、ゲノム、トランスクリプトームから始まる分子の依存関係を、代謝パスウェイへマッピングする。統合型とし、多階層データを同時に扱う枠組みを用いる。また、その一環とし、マウスの知見をヒトのデータに統合し解析する方法を開発する。これらの手法の実証を、メタボローム(馬場)、ゲノム(鈴木)、エピゲノム(伊藤)、トランスクリプトーム(鈴木)、プロテオーム(中山)計測のデータを用いて行っていく。そして、がん転移などの環境適応の解析(中山らと)、炎症疾患(岡田)、糖代謝(黒田)、薬剤応答による動的薬剤選択(松田らと)等のアダプテーション解明と個別化医療へ応用する。
本計画研究では、トランスオミクス解析の手法の提案・開発、ヒトのデータを中心とした解析、そしてマウスの知見をヒトへマッピング・変換、共通する機序を探ることを主軸とする。まず、トランスオミクス解析の方法論として、知識型、因果・階層型、統合型の3種類の方法を検討する。知識型として、染色体構造などのゲノムアノテーションデータにより、ゲノムから代謝にいたる制約を見出すことでフィルタリングを行う。因果・階層型として、ゲノム、トランスクリプトームから始まる分子の依存関係を、代謝パスウェイへマッピングする。統合型とし、多階層データを同時に扱う枠組みを用いる。また、その一環とし、マウスの知見をヒトのデータに統合し解析する方法を開発する。これらの手法の実証を、メタボローム(馬場)、ゲノム(鈴木)、エピゲノム(伊藤)、トランスクリプトーム(鈴木)、プロテオーム(中山)計測のデータを用いて行っていく。そして、がん転移などの環境適応の解析(中山らと)、炎症疾患(岡田)、糖代謝(黒田)、薬剤応答による動的薬剤選択(松田らと)等のアダプテーション解明と個別化医療へ応用する。
参考文献
- Ishigaki, K., Kochi, Y., Suzuki, A., Tsuchida, Y., Tsuchiya, H., Sumitomo, S., Yamaguchi, K., Nagafuchi, Y., Nakachi, S., Kato, R., Sakurai, K., Shoda, H., Ikari, K., Taniguchi, A., Yamanaka, H., Miya, F., Tsunoda, T., Okada, Y., Momozawa, Y., Kamatani, Y., Yamada, R., Kubo, M., Fujio, K., Yamamoto, K. Polygenic burdens on cell-specific pathways underlie the risk of rheumatoid arthritis. Nature Genetics 49, 1120-1125 (2017).
- Fujimoto, A., Furuta, M., Totoki, Y., Tsunoda, T., (equal contribution) Kato, M., Shiraishi, Y., Tanaka, H., Taniguchi, H., Kawakami, Y., Ueno, M., Gotoh, K., Ariizumi, S., Wardell, C. P., Hayami, S., Nakamura, T., Aikata, H., Arihiro, K., Boroevich, K. A., Abe, T., Nakano, K., Maejima, K., Sasaki-Oku, A., Ohsawa, A., Shibuya, T., Nakamura, H., Hama, N., Hosoda, F., Arai, Y., Ohashi, S., Urushidate, T., Nagae, G., Yamamoto, S., Ueda, H., Tatsuno, K., Ojima, H., Hiraoka, N., Okusaka, T., Kubo, M., Marubashi, S., Yamada, T., Hirano, S., Yamamoto, M., Ohdan, H., Shimada, K., Ishikawa, O., Yamaue, H., Chayama, K., Miyano, S., Aburatani, H., Shibata, T. & Nakagawa, H. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nature genetics 48, 500-509 (2016).
- Fujimoto, A., Furuta, M., Shiraishi, Y., Gotoh, K., Kawakami, Y., Arihiro, K., Nakamura, T., Ueno, M., Ariizumi, S., Nguyen, H. H., Shigemizu, D., Abe, T., Boroevich, K. A., Nakano, K., Sasaki, A., Kitada, R., Maejima, K., Yamamoto, Y., Tanaka, H., Shibuya, T., Shibata, T., Ojima, H., Shimada, K., Hayami, S., Shigekawa, Y., Aikata, H., Ohdan, H., Marubashi, S., Yamada, T., Kubo, M., Hirano, S., Ishikawa, O., Yamamoto, M., Yamaue, H., Chayama, K., Miyano, S., Tsunoda, T.* & Nakagawa, H.* (*co-corresponding) Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nature communications 6, 6120 (2015).